
No início, a criança pode experimentar somente um ou dois espasmos por vez, mas, no decorrer de um período de dias ou semanas, estes evoluem para dúzias de espasmos que ocorrem em intervalos de poucos segundos. As convulsões são de difícil controle, e a criança pode chegar a ter mais de 100 convulsões por dia.
Cada espasmo é uma crise epiléptica (ataque epiléptico) composta de uma série de movimentos descontrolados, causados por um excesso de atividade elétrica no cérebro.
Estes ataques foram primeiramente descritos pelo Dr. West (1841) em relação ao seu próprio filho. A Síndrome de West é multifatorial e certos casos pode ter sucetibilidade poligênica ou pode ser completamente ambiental. Algumas crianças podem chorar e/ou gritar antes ou após as convulsões e mostram-se geralmente muito irritadas. O período mais crítico para as convulsões são a hora de dormir ou de acordar, onde a Síndrome apresenta toda a sua face mais cruel.
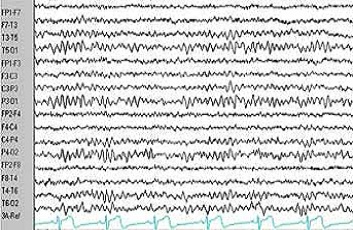
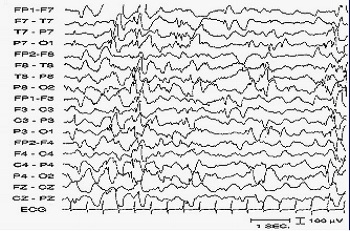
Um eletroencefalograma - EEG pode registrar esta atividade de forma segura e indolor. Um EEG em um bebê com espasmos infantis não apresentam os ritmos brandos e regulares que usualmente se apresenta nessa idade. Ao contrário, ocorrem súbitas eclosões de atividades elétricas, algumas de alto potencial e o registro do EEG aparenta estar caótico. Muitas dessas alterações elétricas tornam-se mais marcantes à medida em que a criança adormece. Este padrão é chamado de "Hypsarritmia" (hypsos=altura e rhytmos=ritmo). É um atraso psicomotor, que pode variar do leve ao severo, dependendo da evolução do caso.
 |  |
As Epilepsias são distúrbios intermitentes das funções do cérebro, frequentemente associados a distúrbios da consciência. O termo é plural pois abrange um enorme grupo de transtornos neurológicos e psiquiátricos. O tipo mais conhecido é o chamado de "Grande Mal", caracterizado por episódios recorrentes de convulsões generalizadas, nas quais o corpo todo estremece numa série de curtos espasmos. Os chamados "ataques" epiléticos variam desde os espasmos, mioclonias, ausências, convulsões febris na infância até os acessos psicomotores em adultos. Atualmente se classificam as convulsões epiléticas em dois grandes grupos: Parciais e Generalizadas.
Sabemos também que, as epilepsias são mudanças bruscas e repentinas no funcionamento bio-elétrico do cérebro e normalmente comparadas a um "curto circuito" momentâneo que afetam as células nervosas (neurônios), fazendo parte de uma disfunção do Sistema Nervoso Central - SNC. Nos "ataques" ou crises epiléticas podem ocorrer perda de consciência momentânea, acompanhada de distúrbios, tais como: abalos musculares, movimentos bruscos, perda do equilíbrio corporal, alterações dos movimentos e das ações de uma pessoa. Posteriormente, há uma pausa, umrepouso devido a exaustão das células nervosas, seguindo um período de confusão mental e/ou sonolência.
AS EPILEPSIAS, ASSIM COMO AS SÍNDROMES RARAS NÃO SÃO CONTAGIOSAS.
Podem estar associadas a quadros de natureza genética, sendo que 70% dos casos são de causas desconhecidas (sem etiologias). Fatores que podem levar a um quadro epilético:
Em casos extremamente graves (quadros epiléticos parciais complexos) existe a possibilidade de uma intervenção neurocirúrgica. Por ser um tratamento invasivo é necessário uma rigorosa avaliação dos riscos e dos benefícios, pois nem todas as crianças estão aptas a uma cirurgia deste tipo.
Atualmente, existem experiências sobre a utilização de Marcapassos Cerebrais que ajudam na organização bio-elétrica do Sistema Nervoso Central - SNC.
Há uma grande melhora dos espasmos infantis com uso intensivo do ACTH (hormônio adrenocorticotrófico) em suas apresentações injetáveis como: ACTHAR (Corticotrophin) ou a sua forma de H.P.ACTHAR Gel (Repository Corticotrophin injection).
Dizemos que este tratamento pode ser heróico e interromper o quadro convulsivo, porém só deve ser utilizado sob rigoroso controle médico e monitoramento cardiopediatrico, já que os corticóides não agem apenas no SNC, mas todo o organismo da criança, inclusive em seu sistema imunológico. Segundo os alguns autores somente se utiliza esta medicação em casos de Síndrome de West considerados criptogênicos, e não em espasmos infantis resultantes de lesões cerebrais, por exemplo. Há casos em que a resposta terapêutica pode aparecer em até 48 ou 72 horas após aplicação de uma primeira dose do ACTH, podendo haver uma possibilidade de recidiva de crises nos casos considerados mais graves na dependência da precocidade do diagnóstico e da extensão e gravidade da lesão cerebral associada.
Outros anticonvulsivantes têm sido utilizados, isoladamente ou em combinação nos casos de espasmo infantis, como o Clonazepam, o Ácido Valpróico, o Fenobarbital e o Vigabatrin.
Tratamento Fisioterápico
Dizemos que este tratamento pode ser heróico e interromper o quadro convulsivo, porém só deve ser utilizado sob rigoroso controle médico e monitoramento cardiopediatrico, já que os corticóides não agem apenas no SNC, mas todo o organismo da criança, inclusive em seu sistema imunológico. Segundo os alguns autores somente se utiliza esta medicação em casos de Síndrome de West considerados criptogênicos, e não em espasmos infantis resultantes de lesões cerebrais, por exemplo. Há casos em que a resposta terapêutica pode aparecer em até 48 ou 72 horas após aplicação de uma primeira dose do ACTH, podendo haver uma possibilidade de recidiva de crises nos casos considerados mais graves na dependência da precocidade do diagnóstico e da extensão e gravidade da lesão cerebral associada.
Outros anticonvulsivantes têm sido utilizados, isoladamente ou em combinação nos casos de espasmo infantis, como o Clonazepam, o Ácido Valpróico, o Fenobarbital e o Vigabatrin.
Em todo paciente com Síndrome de West precisa-se trabalhar primeiramente extensão de cabeça e de tronco, para que depois, então a criança seja estimulada a começar a rolar, arrastar, engatinhar, sentar. Não podemos querer que ela engatinhe, sem que ela consiga fazer extensão cervical. O tratamento deve ser feito seguindo as etapas de evolução, de maturação da criança
Os exercícios fisioterápicos devem obedecer as escalas de maturação. Tendo isso em mente, o fisioterapeuta pode inovar e criar novas maneiras de serem realizados de duas formas: Utilizando a bola coloca-se a criança em DV apoiado com cotovelo em cima da bola, e chama-se a atenção da mesma com um objeto a sua frente.
Deitado no chão, também com um brinquedo à frente.
É importante saber que o tratamento da síndrome de west é igual ao tratamento proposto a criança portadora de paralisia cerebral.
O tratamento fisioterapêutico tem como objetivo principal tratar as seqüelas ou tentar diminuí-las o máximo possível. Como as complicações respiratórias existentes, deve-se fazer fisioterapia respiratória.
É importante saber que o tratamento da síndrome de west é igual ao tratamento proposto a criança portadora de paralisia cerebral.
O tratamento fisioterapêutico tem como objetivo principal tratar as seqüelas ou tentar diminuí-las o máximo possível. Como as complicações respiratórias existentes, deve-se fazer fisioterapia respiratória.
Outro objetivo é tentar-se evitar as deformidades que surgem ou amenizá-las, fazendo-se mobilização passiva e alongamentos. Devido a hipotonia é preciso que se fortaleça os músculos responsáveis pela respiração.
Hidroterapia
Durante a terapia em piscina, o calor da água ajuda a aliviar a espasticidade, mesmo que o alívio seja temporário. Entretanto, a medida que a espasticidade diminui, movimentos passivos podem ser administrados em maior amplitude com menor desconforto para o paciente. Deste modo a amplitude articular pode ser mantida.
Os movimentos passivos devem ser efetuados lentamente e ritmicamente , começando com o tronco e articulações proximais, gradualmente incluindo as articulações distais. Os movimentos devem a princípio ser de natureza oscilatória e a seguir de natureza rotatória. O tronco e os membros devem ser movidos em padrões de movimento com inibição reflexa. O paciente deve respirar profundamente e calmamente, e o momento do estendimento máximo deve coincidir com a expiração. A principal dificuldade em obter uma fixação estável para ambos os pacientes e o terapeuta. Em alguns casos pode ser necessário um segundo fisioterapeuta para ajudar.
Os movimentos passivos devem ser efetuados lentamente e ritmicamente , começando com o tronco e articulações proximais, gradualmente incluindo as articulações distais. Os movimentos devem a princípio ser de natureza oscilatória e a seguir de natureza rotatória. O tronco e os membros devem ser movidos em padrões de movimento com inibição reflexa. O paciente deve respirar profundamente e calmamente, e o momento do estendimento máximo deve coincidir com a expiração. A principal dificuldade em obter uma fixação estável para ambos os pacientes e o terapeuta. Em alguns casos pode ser necessário um segundo fisioterapeuta para ajudar.
Nenhum comentário:
Postar um comentário